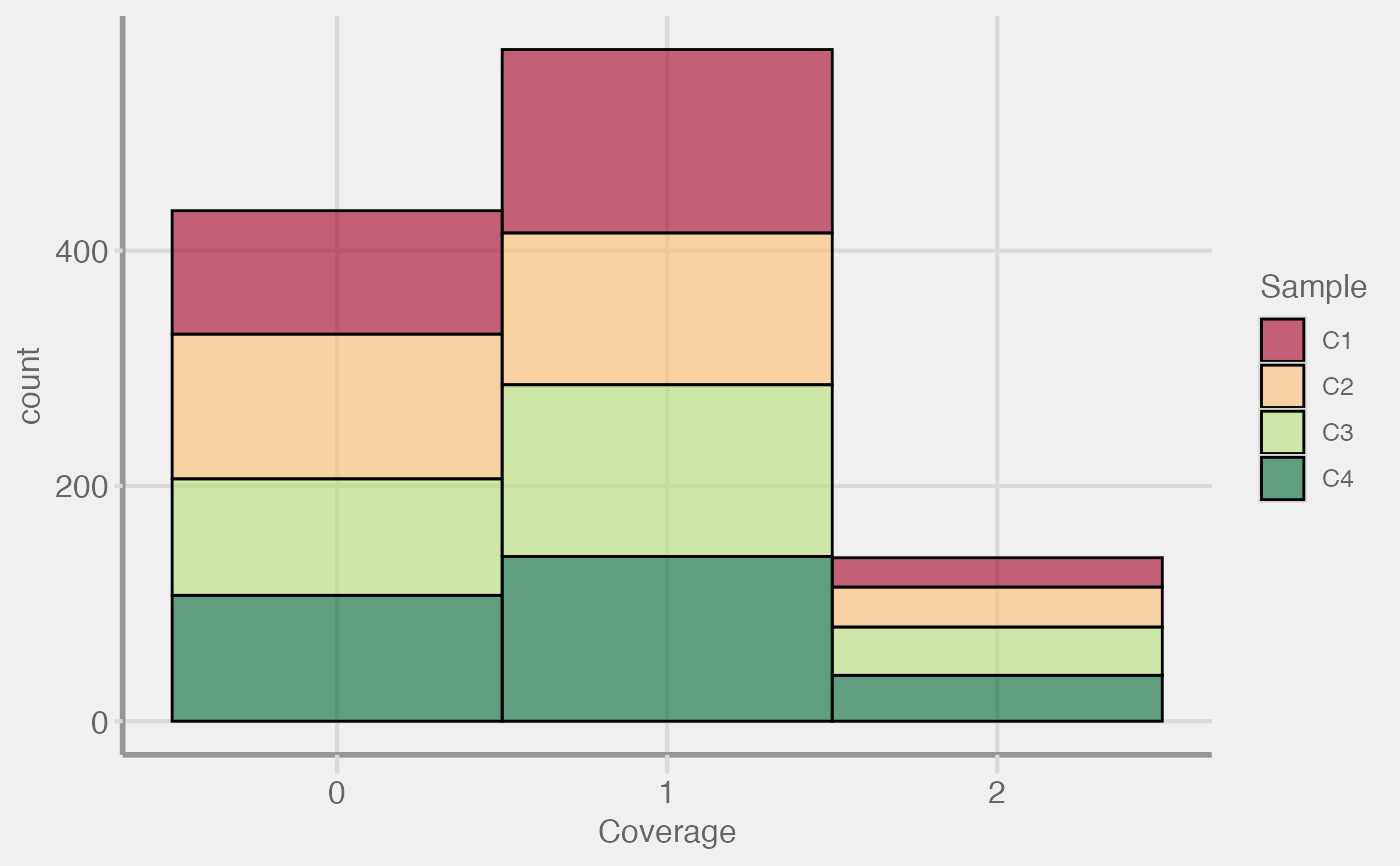
Coverage QC Plots
plot_coverage( scm = NULL, type = c("histogram", "density"), pheno = NULL, max_cov = 100, obs_lim = 1e+06, col_palette = "RdYlGn", show_legend = TRUE, verbose = TRUE )
Arguments
| scm |
|
|---|---|
| type | string; Choose between 'histogram' (histogram) or 'density' (density plot). |
| pheno | string; Col name of colData(m). Will be used as a factor to color different groups |
| max_cov | integer; Maximum coverage value to be plotted. |
| obs_lim | integer; The maximum number of observations (sites*samples) to use. If the dataset is larger that this, random sites will be selected from the genome. |
| col_palette | string; Name of the RColorBrewer palette to use for plotting. |
| show_legend | boolean; Display the legend on the plot |
| verbose | boolean; Flag for outputting function status messages. Default = TRUE |
Value
ggplot2 object
Examples
#>